Nivel 4 - Patologías
El ácido úrico es el producto final de la degradación de las purinas y la hiperuricemia –concentración de urato sérico superior a 7,0 mg/dl en hombres y 6,0 mg/dl en mujeres empleando como método de determinación el ensayo enzimático con uricasa– es el principal condicionante que determina la formación de cristales de urato monosódico.
Leer más: 2.1.1. Hiperuricemia por alteración del metabolismo de purinas. Gota 1 comentario
{kind=link}
La fosforribosilpirofosfato sintetasa, PRPS (E.C. 2.7.6.1), cataliza la síntesis de 5-fosforribosil-1-pirofosfato (PRPP), intermediario en la síntesis de nucleótidos (donde aporta el resto de ribosa de estas moléculas) y de ciertos aminoácidos, en la recuperación de purinas y en el metabolismo del ácido nicotínico.
{kind=link}
La hipoxantina-guanina fosforribosiltransferasa, HPRT (E.C. 2.4.2.8), es la enzima bifuncional de recuperación de hipoxantina y guanina para dar, respectivamente, IMP y GMP.
{kind=link}
Los fallos en dos enzimas del metabolismo de los nucleótidos de purina, adenosina desaminasa (ADA) y purina nucleósido fosforilasa (PNP), son el origen de dos formas de la inmunodeficiencia combinada grave (SCID, “severe combined inmunodeficiency disease”), la enfermedad de los “niños burbuja”.
{kind=link}
En mamíferos, la adenina fosforribosiltransferasa, APRT (E.C. 2.4.2.7), es la única enzima encargada de la recuperación de adenina. Esta enzima se encuentra en todos los tejidos, aunque con mayor concentración en hígado; es una proteína citosólica y homodimérica con 179 residuos de aminoácidos y una masa de 19,5 kDa por subunidad; su gen está en la región cromosómica 16q24.3, tiene 2,8 kb y está formado por 5 exones y 4 intrones.

La deficiencia en APRT (OMIM 102600) determina la incapacidad de producir AMP a partir de adenina con lo que ésta es oxidada por la xantina oxidorreductasa a 2,8-dihidroxiadenina (2,8-DHA). La adenina y la 2,8-DHA se excretan vía renal, pero esta última molécula es extremadamente insoluble y su acumulación da lugar a la formación de piedras que producen cristaluria y litiasis. Los síntomas clínicos son: cólicos renales, hematuria, infecciones urinarias y disuria.
La edad de comienzo de la enfermedad es muy variable; el defecto se hereda de forma autosómica recesiva pero el 60% de los casos descritos corresponde a varones. Los heterocigotos no suelen mostrar anomalías clínicas o bioquímicas. Para los homocigotos se han identificado dos tipos: el tipo I, que predomina en caucásicos, tiene una actividad de la APRT prácticamente indetectable en lisados de eritrocitos; el tipo II tiene del 10-25% de actividad APRT y sólo se ha manifestado en japoneses. Parece que en el tipo II lo que se produce es una menor afinidad de la enzima por el PRPP lo que hace aumentar su KM unas diez veces. En pacientes no japoneses, se han detectado unas 15 mutaciones en el gen de la APRT que se asocian con esta disfunción renal.
Ha sido frecuente la confusión de los cálculos de 2,8-DHA con cálculos de ácido úrico debido a que tienen idéntica reactividad química; la identificación correcta de la 2,8-DHA requiere técnicas más complejas (espectro infrarrojo y otras). Aparte de la detección de 2,8-DHA en la orina, el diagnóstico en el tipo I se confirma con la ausencia de APRT funcional en eritrocitos intactos; en el tipo II se recomiendan estudios genéticos pero también se hacen estudios de resistencia a 2,6-diaminopurina en cultivos de células T o de incorporación de adenina radiactiva en eritrocitos.
El tratamiento de esta deficiencia enzimática incluye la restricción de las purinas de la dieta y un alto consumo de líquido. El alopurinol también ayuda a prevenir la excreción de 2,8-DHA y la formación de piedras.
Colaboración de:
Prof. I. Carrero Ayuso
(Dr. en CC. Biológicas)
Dpto. de Bioq. y Biol. molec. y Fisiol.
E.U. de Fisioterapia
Universidad de Valladolid
{kind=link}
La xantina oxidorreductasa, XOR (E.C. 1.17.3.2), cataliza la oxidación de hipoxantina a xantina y de xantina a ácido úrico en la degradación de purinas.
{kind=link}
La adenilosuccinasa o adenilosuccinato liasa, ADSL (E.C. 4.3.2.2), cataliza el octavo paso en la síntesis de novo de purinas, en el cual el SAICAR (N-succinil-5-aminoimidazol-4-carboxamida ribonucleótido) se transforma en AICAR (5-aminoimidazol-4-carboxamida ribonucleótido),
{kind=link}
En 2004 se describió este defecto hereditario (OMIM 608688) que afecta a la síntesis de novo de purinas y en el que se produce la excreción urinaria masiva de AICA (5-amino-4-imidazolcarboxamida) ribósido en una paciente que también presenta características dismórficas, defectos neurológicos graves y ceguera congénita.
{kind=link}
La adenilato desaminasa (AMP desaminasa, AMPD, E.C. 3.5.4.6) cataliza la conversión del AMP a IMP y amoniaco; en la mayor parte de los tejidos, excepto en el corazón, la ruta predominante del catabolismo del AMP es su desaminación mediante esta enzima.
2.1.8.1. Deficiencia en desoxiguanosina quinasa.
La desoxiguanosina quinasa, dGK (E.C. 2.7.1.113), fosforila los desoxinucleósidos de guanina y sus análogos utilizando ATP y UTP como dadores del grupo fosforilo. La dGK se expresa constitutivamente y se ha encontrado expresión similar en tejido linfoide, bazo, piel, hígado y cerebro.
{kind=link}
Colaboración de :
Dra. Isabel Carrero Ayuso
Área de Bioquímica y Biología Molecular
E.U. de Fisioterapia de Soria
Universidad de Valladolid
E-mail:
En este apartado se tratan algunas de las alteraciones relacionadas con el consumo de pirimidinas, más concretamente trataremos los siguientes trastornos:

- 2.2.1. Aciduria orótica hereditaria.
- 2.2.2. Alteraciones en actividades 5’-nucleotidasa.
- 2.2.2.1. Deficiencia en pirimidina 5’-nucleotidasa.
- 2.2.2.2. Superactividad 5’-nucleotidasa.
- 2.2.3. Deficiencia en dihidropirimidina deshidrogenasa.
- 2.2.4. Deficiencia en dihidropirimidinasa.
- 2.2.5. Deficiencia en ureidopropionasa.
- 2.2.6. Defectos en enzimas mitocondriales.
- 2.2.6.1. Deficiencia en timidina fosforilasa.
- 2.2.6.2. Deficiencia en timidina quinasa-2.
Colaboración de:
Prof. I. Carrero Ayuso
(Dr. en CC. Biológicas)
Dpto. de Bioq. y Biol. molec. y Fisiol.
E.U. de Fisioterapia
Universidad de Valladolid
{kind=link}
Hay varios defectos hereditarios que conducen a la excreción de elevadas cantidades de ácido orótico en orina; el término aciduria orótica hereditaria se aplica a la deficiencia en UMP sintasa (UMPS, enzima bifuncional de la síntesis de novo de pirimidinas que incluye las actividades orotato fosforribosiltransferasa, OPRT, E.C. 2.4.2.10, y orotidilato descarboxilasa, OMPD, E.C. 4.1.1.23).
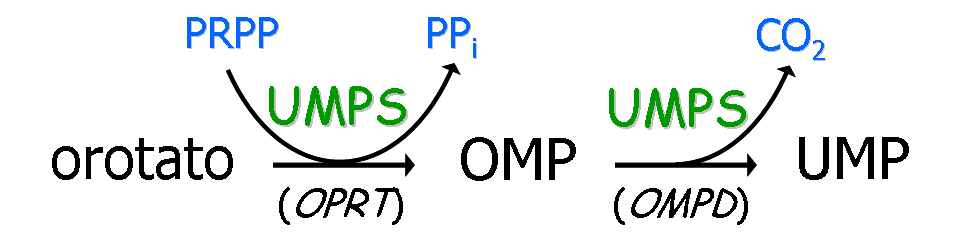
Esta deficiencia es un raro defecto genético de tipo autosómico recesivo en el que los niños afectados no pueden sintetizar nucleótidos pirimidínicos y excretan grandes cantidades de orotato en orina produciéndose cristaluria en el tracto urinario y, sobre todo, en la orina una vez excretada; hay también obstrucciones uretrales y ureterales, hematuria y azotemia, todo ello acompañado de retraso mental, retraso en el crecimiento, anemia megaloblástica y leucopenia, síntomas que pueden revertirse administrando desde las primeras semanas de vida uridina o citidina con lo que, aparte de contrarrestar la deficiencia en las dos enzimas citadas, se producirá UTP que actuará como retroinhibidor de la carbamoil-P sintetasa II y hará que se reduzca la síntesis de orotato.
Sin tratamiento, el déficit de nucleótidos con uracilo hace que disminuyan los niveles de ciertos UDP-azúcares que son necesarios para la utilización de galactosa y para la síntesis de glucógeno, y el de nucleótidos de citosina hace que disminuyan los niveles de CDP-etanolamina y CDP-colina que son intermediarios en la síntesis de fosfolípidos; estas carencias pueden ser la base de debilidad en el músculo esquelético.
El gen de la UMPS está en el cromosoma 3q13 y contiene 6 exones. La frecuencia génica de la deficiencia en UMPS es bastante alta (1/200-1/6000) lo que no se corresponde con el bajo número de casos documentados. Se ha sugerido que los pacientes que sobreviven constituyen una variante menos grave de las formas más severas que son letales in utero.
En la mayoría de los casos, aciduria orótica tipo 1 (OMIM 258900), se han detectado mutaciones puntuales en el dominio OPRT; cuando está afectada la actividad OMPD se habla de aciduria orótica tipo 2 (OMIM 258920), en este caso hay anomalías neurológicas y orotidinuria pero no anemia megaloblástica.
Aparte de los citados, la aciduria orótica puede tener otros orígenes siendo el caso más destacado el de los defectos en el ciclo de la urea; p.e., el déficit en ornitina transcarbamoilasa produce la acumulación de carbamoil-P que se derivará a la síntesis de pirimidinas. Esta sería la aciduria orótica tipo 3.
Colaboración de:
Prof. I. Carrero Ayuso
(Dr. en CC. Biológicas)
Dpto. de Bioq. y Biol. molec. y Fisiol.
E.U. de Fisioterapia
Universidad de Valladolid
Las 5’-nucleotidasas (E.C. 3.1.3.5) catalizan la desfosforilación de nucleósidos 5’-monofosfato a sus correspondientes nucleósidos. Existen siete 5’-nucleotidasas con especificidad de sustrato, distribución, localización celular, regulación y estructura primaria diferentes; hay una forma ectosólica (eN) unida a membrana que está implicada en la señalización extracelular y otras seis intracelulares, cinco de ellas citosólicas (cN-Ia y b, cN-II, cN-III y cdN) y una mitocondrial (mdN).
2.2.2.1. Deficiencia en pirimidina 5’-nucleotidasa.
Aunque su expresión es ubicua, en los glóbulos rojos hay dos tipos de 5’-nucleotidasas que actúan sobre pirimidinas, la primera, también denominada UMP hidrolasa 1 (UMPH-1), es activa con ribonucleótidos y la segunda (UMPH-2) no es tan específica de pirimidinas y es eficiente con desoxirribonucleótidos. La mayor parte de la actividad reside en la UMPH-1 y cuando hay déficit de esta enzima se acumulan nucleótidos de pirimidina (CTP y UTP) en los eritrocitos; esta acumulación altera el metabolismo de los glóbulos rojos provocando aumento de los niveles de CDP-colina y CDP-etanolamina, secuestro de Mg2+, reducción de la actividad PRPS, fallos en la ruta de las pentosas fosfato o aumento de los niveles de glutatión reducido.
La UMPH-1 es citosólica –la 5’-nucleotidasa citosólica III (cNIII)–, tiene 286 aminoácidos, su masa es 34 kDa y es monomérica; cataliza la desfosforilación de UMP y CMP. Esta enzima requiere Mg2+ y es inhibida por AMP, algunas bases púricas, nucleósidos de purina y pirimidina, cationes divalentes o metales pesados y por agentes activos contra grupos sufhidrilo. El gen de la UMPH-1 está en 7p14-15, tiene 50 kb, 11 exones y genera tres ARNm distintos. En la deficiencia en pirimidina 5’-nucleotidasa se han encontrado unas veinte mutaciones distintas en el gen de esta enzima
La deficiencia en pirimidina-5' nucleotidasa es un defecto autosómico recesivo que da lugar a anemia hemolítica no esferocítica (OMIM 266120); la ictericia y la esplenomegalia suelen ser comunes y algunas veces hay hepatomegalia. En algunos pacientes se dan distintos grados de retraso en el desarrollo y dificultades en el aprendizaje.
Para el diagnóstico, es útil determinar la relación nucleótidos de purina/nucleótidos de pirimidina en los glóbulos rojos a partir del cociente de absorbancias A260/A280; en la deficiencia en pirimidina 5’-nucleotidasa este cociente da un valor por debajo de lo normal.
2.2.2.2. Superactividad 5’-nucleotidasa.
En el síndrome relacionado con el autismo denominado desorden del desarrollo asociado con incremento de la actividad de la nucleotidasa (NAPDD, “nucleotidase associated pervasive developmental disorder”) aumenta hasta 10 veces la actividad de una 5’-nucleotidasa de purinas y pirimidinas. Los individuos afectados presentan síntomas neurológicos como ataques, ataxia, andar torpe, daños en el control motor fino o retraso en el lenguaje y su comportamiento muestra hiperactividad, carácter impulsivo, poca capacidad de atención, escasa interacción social y agresividad. Parece que este defecto se podría relacionar con la 5’-nucleotidasa unida a membrana.
En glóbulos rojos y fibroblastos de pacientes con LNS (ver en N4-2.1.1.2.) se ha detectado también actividad aumentada de la 5’-nucleotidasa citosólica cN-II, una enzima muy extendida y específica para IMP, GMP y sus derivados desoxi. Esta superactividad de la cN-II se relacionaría con la patogenia de los síntomas neurológicos del LNS a través de la generación de AICA ribósido a partir de AICAR.
Colaboración de:
Prof. I. Carrero Ayuso
(Dr. en CC. Biológicas)
Dpto. de Bioq. y Biol. molec. y Fisiol.
E.U. de Fisioterapia
Universidad de Valladolid
{kind=link}
Existen deficiencias en la dihidropirimidina deshidrogenasa, DPD (E.C. 1.3.1.2), enzima homodimérica que cataliza el paso limitante de la degradación de pirimidinas (paso de uracilo y timina a dihidrouracilo y dihidrotimina, respectivamente).

La DPD se encuentra en la mayor parte de los tejidos con actividad más elevada en linfocitos. Puede tener isoenzimas; la forma hepática es una flavoenzima ferrosulfurada y homodimérica.
Cuando existe este déficit (OMIM 274270), en la orina se encuentran niveles elevados de uracilo, timina y 5-hidroximetiluracilo. Características clínicas son: retraso mental y en el desarrollo, espasticidad, y ataques; la epilepsia podría ser una característica común a todos los afectados, aunque hay muchos pacientes asintomáticos. También se han encontrado efectos tóxicos inesperados en pacientes con este déficit enzimático y cáncer al ser tratados con dosis habituales del agente quimioterapéutico 5-fluorouracilo.
El gen de la DPD está en la región cromosómica 1p22. Los pacientes con síntomas clínicos de esta deficiencia son homocigóticos; los que presentan toxicidad ante el 5-fluoruracilo pueden ser homo o heterocigóticos.
El diagnóstico se basa en la aparición de mayores concentraciones de uracilo y timina en orina, pero hay que hacer ensayos enzimáticos porque la uraciluria y la timinuria también pueden aparecer en otros trastornos como la dihidropirimidinuria y en defectos del ciclo de la urea.
Colaboración de:
Prof. I. Carrero Ayuso
(Dr. en CC. Biológicas)
Dpto. de Bioq. y Biol. molec. y Fisiol.
E.U. de Fisioterapia
Universidad de Valladolid
{kind=link}
La dihidropirimidinasa, DHP (E.C. 3.5.2.2), cataliza la hidrólisis de los anillos de dihidrouracilo y dihidrotimina para dar sus correspondientes derivados no cíclicos.
{kind=link}
La b-ureidopropionasa, UP (E.C. 3.5.1.6), cataliza la conversión de los ácidos ureidopropiónico y ureidoisobutírico en b-alanina y ácido b-aminoisobutirato, respectivamente.
{kind=link}
2.2.6.1. Deficiencia en timidina fosforilasa.
La timidina fosforilasa, TP (E.C. 2.4.2.4), cataliza el paso de (desoxi)timidina a timina.
2.3. ABREVIATURAS
Estas son las abreviaturas usadas en esta serie de artículos sobre los trastornos relacionados con Purinas y Pirimidinas
3. Trastornos relacionados con los aminoácidos
Trastornos del metabolismo de los aminoácidos
{kind=link}
La fenilcetonuria es un trastorno que se caracteriza porque la conversión de la fenilalanina en tirosina es defectuosa.
Trastorno metabólico raro, en el que se excreta el ácido p-hidroxifenilpirúvico. Los pacientes con afectación hepática excretan cantidades anormales de este cetoácido, pero las concentraciones halladas en la tirosinosis eran cerca de 10 veces mayores que los valores más elevados observados en las enfermedades hepáticas.
{kind=link}
Se trata de un error metabólico congénito. Es una enfermedad poco frecuente en la que el ácido homogentísico, un intermediario de la ruta catabólica de la fenilalanina, no puede metabolizarse provocando aciduria (orina negra), ocronosis (pigmentación del tejido conjuntivo) y artrosis.
{kind=link}
El albinismo es un defecto metabólico hereditario, limitado a las células pigmentarias, los melanocitos. Una deficiencia de tirosinasas origina la incapacidad de formar melanina.

 Este proyecto ha sido subvencionado parcialmente por el Ministerio de Ciencia y Tecnología, Programa de Fomento de la Investigación Técnica del Plan Nacional de Investigación Científica, Desarrollo e Innovación Tecnológica.Dirección técnica y desarrollo: CMP Centro de Microinformática y Programación SRL
Este proyecto ha sido subvencionado parcialmente por el Ministerio de Ciencia y Tecnología, Programa de Fomento de la Investigación Técnica del Plan Nacional de Investigación Científica, Desarrollo e Innovación Tecnológica.Dirección técnica y desarrollo: CMP Centro de Microinformática y Programación SRLNovedad
Modelo bidireccional y triestratificado
Autor: Profesor G. Gómez-Jarabo
Director de biopsicologia.net
Desarrollo técnico: CMP Centro de Microinformática y Programación SRL
Dirección técnica: Emilio Garijo Soler